
ABSTRAK
Lebih dari seabad yang lalu, diketahui bahwa akumulasi agregat protein yang teratur, fibril amiloid, menyertai beberapa patologi serius dan sebagian besar masih tidak dapat disembuhkan, termasuk penyakit Alzheimer dan Parkinson. Kesenjangan yang mencolok antara penelitian selama beberapa dekade yang mengidentifikasi amiloid sebagai salah satu pendorong utama neurodegenerasi dan kurangnya terapi anti-amiloid yang efektif mengungkapkan kontradiksi yang membingungkan, yang kami definisikan sebagai “paradoks amiloid.” Untuk mengatasi paradoks ini, di sini kami merangkum dan menganalisis perspektif terkini tentang sifat unik dan mekanisme patogenik amiloid, menyoroti variabilitas dan kompleksitas konsekuensi biologisnya dan mengungkap risiko dan keterbatasan yang dihadapi dalam memerangi agregat ini. Kami mengonseptualisasikan patogenisitas fibril amiloid sebagai kaskade kompleks yang meluas jauh melampaui sitotoksisitas langsung, seperti yang timbul dari gangguan membran dan organel seluler lainnya. Tinjauan ini mencakup efek disruptif amiloid pada proses seluler dan kemampuan untuk memicu respons inflamasi, ketahanannya terhadap degradasi, kapasitas untuk beregenerasi setelah kerusakan yang nyata, kecenderungan untuk menyebar ke seluruh organisme, kecenderungan untuk transformasi yang meningkatkan sitotoksisitas, dan kemampuan untuk mengisolasi dan memodifikasi biomolekul esensial secara patologis. Analisis terpadu ini mengungkap mengapa pendekatan terapi target tunggal sering gagal dan menunjukkan bahwa strategi anti-amiloid yang efektif harus mengatasi berbagai aspek patogenisitas amiloid secara bersamaan. Pembingkaian ulang konseptual dari ancaman fibril amiloid membantu menjelaskan asal-usul paradoks amiloid, meningkatkan pemahaman kita tentang agen patogen yang kompleks ini, dan memberikan dasar untuk mengembangkan strategi terapi yang lebih efektif dan aman untuk penyakit neurodegeneratif. Strategi ini harus mengatasi sifat patogenisitas amiloid yang kompleks dan saling berhubungan daripada menargetkan aspek-aspek yang terisolasi.
Singkatan
Penyakit Alzheimer
sklerosis lateral amiotrofik
Bahasa Indonesia: ARIA
kelainan pencitraan terkait amiloid
Bahasa Indonesia: ATP
adenosin trifosfat
Sebuah
β-peptida amiloid
BBB
penghalang darah-otak
BRIKOS
Bri2, kondromodulin-1, dan protein pro-surfaktan C
gas alam cair
sintase siklik GMP-AMP
DAMP (basah kuyup)
pola molekuler terkait kerusakan
DNA
asam deoksiribonukleat
UGD
retikulum endoplasma
GAPDH
gliseraldehida-3-fosfat dehidrogenase
HSPG (Honours of Life) adalah istilah umum untuk istilah yang digunakan untuk menggambarkan situasi di mana seseorang tidak dapat melihat sesuatu yang baru.
proteoglikan heparan sulfat
HTRA1
persyaratan suhu tinggi A serine peptidase 1
Aplikasi IAPP
polipeptida amiloid pulau
IRF3
faktor pengatur interferon 3
LAG3
gen aktivasi limfosit 3
mitoBK Ca
saluran kalium yang diaktifkan kalsium dengan konduktansi besar mitokondria
MMP
matriks metaloproteinase
DNA mt
DNA mitokondria
NADPH
nikotinamida adenin dinukleotida fosfat
NLR
Reseptor mirip NOD
PROTAC (Produk Anti Penipuan)
chimera yang menargetkan proteolisis
PRR
reseptor pengenalan pola
RME
endositosis yang diperantarai reseptor
RNA
asam ribonukleat
ROS
spesies oksigen reaktif
SOD1
superoksida dismutase 1
MENYENGAT
stimulator protein gen interferon
TBK1
Kinase pengikat TANK 1
TLR
reseptor tipe tol
TNT
nanotube terowongan
UPR
respons protein yang tidak terlipat
1 Pendahuluan
Fibril amiloid adalah agregat protein yang sangat teratur yang terdiri dari banyak untai-β yang ditumpuk sejajar satu sama lain dan tegak lurus terhadap sumbu panjang serat (struktur β silang), yang memberikan stabilitas tinggi mereka [ 1 – 5 ]. Selama bertahun-tahun, bentuk protein agregat ini kurang mendapat perhatian. Namun, ini berubah secara signifikan dengan penemuan bahwa pembentukan dan akumulasi fibril amiloid dalam tubuh manusia (proses yang dikenal sebagai amiloidosis) dikaitkan dengan kondisi patologis yang parah [ 6 , 7 ]. Kasus pertama yang terdokumentasi dari plak amiloid di otak pasien terjadi pada awal abad ke-20 [ 8 ]. Meskipun demikian, peran akumulasi amiloid dalam tubuh masih kurang dipahami selama beberapa dekade [ 9 , 10 ]. Pada tahun 1984, para peneliti pertama kali mengusulkan bahwa agregasi β-peptida (Aβ) amiloid dapat menjadi penyebab utama penyakit Alzheimer [ 11 ]. Hipotesis ini awalnya menghadapi skeptisisme dalam komunitas ilmiah, tetapi 7 tahun kemudian, apa yang disebut “hipotesis amiloid” dari patogenesis penyakit Alzheimer ditetapkan. Hipotesis ini segera memperoleh pengakuan sebagai paradigma yang menentukan dalam studi penyakit neurodegeneratif, termasuk penyakit Parkinson, Huntington, dan Creutzfeldt–Jakob [ 12 ]. Awal abad ke-21 menandai penemuan amiloid fungsional di seluruh organisme [ 13 – 15 ], dari bakteri hingga manusia. Amiloid fungsional, seperti curli bakteri dan hidrofobin jamur, ditemukan memainkan peran penting dalam proses alami, termasuk pembentukan biofilm, pensinyalan, dan integritas struktural. Pada mamalia, amiloid fungsional terlibat dalam berbagai proses fisiologis, termasuk produksi melanin, persistensi memori, pembentukan trombosit, nekrosis, reproduksi, penyimpanan hormon, dan aktivitas antimikroba [ 16 , 17 ]. Meskipun demikian, dan juga mempertimbangkan ide-ide yang muncul tentang faktor-faktor tambahan yang terlibat dalam patogenesis penyakit neurodegeneratif, seperti disregulasi ion logam dan neuroinflamasi kronis [ 18 ], serta bukti sitotoksisitas tinggi prekursor amiloid [ 19 , 20 ], fibril amiloid dianggap dan masih dianggap sebagai penyebab utama patologi ini. Lebih jauh, menjadi jelas bahwa akumulasi fibril amiloid tidak eksklusif untuk penyakit neurodegeneratif tetapi merupakan karakteristik dari banyak patologi lokal dan sistemik lainnya. Sampai saat ini, beberapa lusin protein amiloidogenik telah diidentifikasi, yang mengarah pada penemuan lebih banyak penyakit, karena agregasi protein yang sama dapat berkontribusi pada perkembangan beberapa patologi [ 2 ,21 , 22 ].
Statistik saat ini menggarisbawahi prevalensi penyakit terkait amiloid. Penyakit Alzheimer, sering disebut sebagai “epidemi abad ke-21,” adalah bentuk demensia yang paling umum, mencakup 60%–70% dari semua kasus dan mempengaruhi lebih dari 55 juta orang di seluruh dunia [ 23 – 25 ]. Demensia saat ini menempati peringkat ketujuh penyebab kematian secara global [ 23 ]. Pada tahun 2050, jumlah individu yang didiagnosis dengan demensia dan penyakit Alzheimer diperkirakan akan mencapai 139 juta [ 26 , 27 ]. Demikian pula, prevalensi penyakit Parkinson melampaui 8,5 juta secara global pada tahun 2019, dengan perkiraan menunjukkan peningkatan menjadi 17 juta pada tahun 2040 [ 26 , 28 ]. Penyakit Huntington, meskipun lebih langka daripada Alzheimer atau Parkinson, juga menunjukkan tren peningkatan yang mengkhawatirkan. Perkiraan global dari beberapa dekade terakhir menempatkan prevalensinya pada sekitar 120 ribu orang di populasi Barat [ 29 ], dan jumlah ini diproyeksikan akan meningkat secara signifikan [ 30 ]. Meskipun prevalensi penyakit neurodegeneratif yang terkait dengan amiloidosis dapat bervariasi berdasarkan wilayah dan faktor demografi, tren keseluruhan menunjukkan peningkatan yang mengerikan dalam patologi ini.
Meskipun patologi terkait amiloid sangat parah dan umum, pengobatan yang efektif masih belum tersedia untuk sebagian besar penyakit ini. Meskipun hasil uji klinis yang melibatkan berbagai antibodi anti-amiloid untuk pengobatan penyakit Alzheimer dan Parkinson menjanjikan [ 31 , 32 ], belum ada yang mengarah pada pemulihan pasien yang efektif dan aman. Karena beberapa perspektif menunjukkan bahwa fibril amiloid mungkin merupakan fenomena sekunder pada penyakit neurodegeneratif, yang menghubungkan kausalitas primer dengan faktor -faktor seperti disregulasi ion logam, neuroinflamasi kronis, atau sitotoksisitas oligomer amiloid [ 18–20 , 33 ], kegagalan terapi pada neurodegenerasi sering dikaitkan dengan kesalahan identifikasi target terapi [ 34 , 35]]. Namun, dalam tinjauan ini, kami mempertimbangkan kemungkinan penyebab lain dari kesenjangan antara penelitian selama beberapa dekade yang mengidentifikasi fibril amiloid sebagai salah satu pendorong utama neurodegenerasi dan kurangnya terapi anti-amiloid yang efektif yang mengungkap kontradiksi yang membingungkan, yang kami definisikan sebagai “paradoks amiloid.” Kami percaya bahwa asal mula paradoks ini terletak pada sifat-sifat agregat protein ini. Sifat-sifat unik ini menciptakan tantangan yang saat ini tidak dapat diatasi untuk memeranginya secara efektif dan aman. Mengingat hal ini, untuk mengatasi paradoks ini, kami merangkum dan menganalisis perspektif terkini tentang sifat-sifat unik dan mekanisme patogenik fibril amiloid yang berkontribusi pada tantangan terapeutik ini. Secara khusus, kami memeriksa sifat patogenisitas amiloid yang multifaset, menganalisis bagaimana fibril mengganggu proses seluler fundamental dan integritas organel di berbagai tingkatan. Lebih jauh, kami mengeksplorasi mekanisme yang digunakan fibril untuk memicu dan mempertahankan respons inflamasi, yang berkontribusi pada kerusakan jaringan kronis. Kami juga merinci faktor-faktor yang memberikan stabilitas tinggi pada fibril, ketahanannya terhadap degradasi lengkap, dan kapasitasnya yang luar biasa untuk beregenerasi dari fragmen, di samping membahas jalur yang memungkinkan penyebaran patologi amiloid ke seluruh organisme. Selain itu, tinjauan ini mempertimbangkan implikasi polimorfisme fibril, kemampuannya untuk menjalani transformasi struktural, dan penyerapan biomolekul vital lainnya, yang dapat memodulasi efek patogenik fibril amiloid. Dengan meringkas pengetahuan terkini tentang berbagai aspek ini, tinjauan ini bertujuan untuk memberikan pemahaman yang komprehensif tentang kompleksitas yang melekat dalam memerangi patologi amiloid. Pembingkaian ulang konseptual tentang ancaman fibril amiloid tidak hanya membantu menjelaskan asal-usul “paradoks amiloid”, tetapi juga memfasilitasi pengembangan strategi terapi yang lebih efektif dan aman untuk penyakit neurodegeneratif dan amiloidosis lainnya. Strategi tersebut harus memperhitungkan sifat patobiologi amiloid yang rumit dan saling terkait serta potensi keterbatasan atau efek samping dari intervensi terapi target tunggal.
2 Mekanisme Patogenisitas Amiloid
2.1 “Sabotase”: Gangguan Proses Seluler Fundamental di Berbagai Tingkat
Meskipun amiloid terutama terletak di luar sel, jalur masuknya ke dalam sel telah diidentifikasi [ 36 – 41 ]. Akibatnya, fibril amiloid dapat menyebabkan disfungsi sel dan kematian melalui berbagai mekanisme, yang menggarisbawahi potensi patologisnya yang tinggi. Aspek utama dari efek patogeniknya melibatkan gangguan integritas membran, disfungsi organel, dan induksi stres oksidatif. Di sini kami memeriksa masing-masing mekanisme ini secara rinci dan membahas interkoneksinya. Karena amiloid tidak hanya memengaruhi sel-sel otak tetapi juga sel-sel organ lain [ 42 ], narasi selanjutnya akan cukup untuk sel-sel dari berbagai organ dan jaringan di seluruh tubuh.
Gangguan integritas membran plasma merupakan pendorong utama sitotoksisitas fibril amiloid. (Gambar 1 ). Saat ini, dua mekanisme interaksi fibril amiloid–membran sedang diselidiki. Beberapa penelitian menunjukkan bahwa hanya lapisan bulu yang membungkus inti fibril yang membentuk interaksi kuat dengan membran [ 43 , 44 ]. Penelitian lain menunjukkan bahwa ujung fibril amiloid, yang bertindak seperti jarum mikroskopis, dapat menyerang lapisan lipid, sehingga menyebabkan kerusakan membran [ 38 , 45-50 ]. Interaksi antara fibril amiloid dan membran plasma ini dapat memulai kematian sel apoptosis [ 38 , 51 , 52 ]. Lebih jauh lagi, interaksi ini menyebabkan gangguan homeostasis Ca2⁺ karena pembentukan pori-pori dan saluran amiloid yang meningkatkan permeabilitas membran [ 46 , 53 ] (Gambar 2 ).
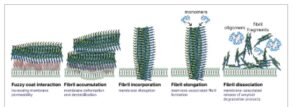
GAMBAR 1
Buka di penampil gambar
Presentasi PowerPoint
Varian interaksi fibril amiloid–membran. Fibril amiloid berinteraksi dengan membran sel melalui beberapa mekanisme, termasuk asosiasi permukaan melalui lapisan bulu halus, serta penggabungan, pemanjangan, akumulasi, dan disosiasi fibril. Interaksi ini berkontribusi terhadap destabilisasi membran, peningkatan permeabilitas, kerusakan struktural, dan disfungsi sel.
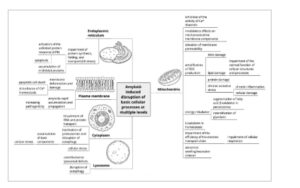
GAMBAR 2
Buka di penampil gambar
Presentasi PowerPoint
Ilustrasi skematis dampak patologis fibril amiloid pada organel sel. Gambar ini menyoroti efek merugikan fibril amiloid pada berbagai struktur sel, termasuk membran plasma, endosom, lisosom, mitokondria, dan retikulum endoplasma. Gangguan pada tingkat organel individu tidak terjadi secara terpisah, tetapi saling memperkuat, menciptakan respons stres berjenjang pada sel. Hal ini terlibat dalam disfungsi sel, yang berkontribusi terhadap perkembangan amiloidosis dan penyakit terkaitnya.
Selain itu, bukti menunjukkan bahwa disosiasi fibril yang dimediasi membran menghasilkan pelepasan oligomer, yang dapat berkontribusi terhadap toksisitas amiloid [ 54 – 56 ]. Kerusakan membran tidak hanya disebabkan oleh interaksi langsung dengan amiloid dewasa tetapi juga oleh agregasi yang teratur dan tidak teratur pada permukaan membran, yang disebabkan oleh akumulasi protein dan/atau fragmen fibril yang terkait dengan membran [ 38 , 49 , 57 – 61 ]. Studi pada α-synuclein telah menunjukkan bahwa agregasi tersebut pada permukaan membran dapat menginduksi penyebaran agregat amiloid yang cepat. Proses ini juga melibatkan berbagai mekanisme masuknya fibril ke dalam sel dan transmisinya antar sel melalui eksositosis [ 62 , 63 ] dan tunneling nanotube (TNT) [ 64 – 66 ].
Kisaran target fibril amiloid meluas melampaui membran plasma. Organel intraseluler juga dipengaruhi oleh agregat toksik ini, yang menyebabkan disfungsi seluler yang meluas. Di antara organel ini adalah mitokondria (Gambar 2 ), yang memainkan peran sentral dalam menjaga metabolisme energi otak dan terkait erat dengan patogenesis penyakit neurodegeneratif [ 67 ]. Beberapa penelitian telah menunjukkan bahwa fibril amiloid mampu berinteraksi dengan membran mitokondria, menyebabkan berbagai bentuk kerusakan dan disfungsi mitokondria. Misalnya, interaksi antara fibril Aβ dan membran mitokondria menghambat aktivitas saluran Ca 2 ⁺ (mitoBK Ca ) [ 68 ], menunjukkan efek modulasi pada komponen mekanosensitif membran mitokondria dan berkontribusi pada perkembangan penyakit Alzheimer. Lebih jauh lagi, paparan mitokondria otak yang terisolasi mengubah permeabilitas membran mitokondria mereka dan menyebabkan peningkatan yang kuat dalam produksi spesies oksigen reaktif (ROS) dalam kasus paparan fibril amiloid α-synuclein [ 69 ], sedangkan dalam kasus fibril amiloid insulin, hal itu hanya menghasilkan peningkatan sedang dalam pelepasan enzim mitokondria [ 69 , 70 ]. Studi-studi ini menunjukkan korelasi antara efek toksik amiloid dan interaksinya dengan membran melalui kontak nonspesifik (melalui daerah hidrofobik dan elektrostatik pada permukaan fibril) dan spesifik (dengan komponen membran tertentu). Fibril amiloid superoksida dismutase 1 (SOD1) juga menginduksi kerusakan mitokondria, memicu neuroinflamasi, dan mengaktifkan sel-sel mikroglia, yang mendorong neurodegenerasi pada sklerosis lateral amiotrofik (ALS) [ 71 ].
Penelitian menunjukkan bahwa interaksi antara fibril amiloid dan membran mitokondria tidak hanya menyebabkan kerusakan yang disebutkan di atas tetapi juga mengganggu efisiensi rantai transpor elektron [ 72 , 73 ]. Secara khusus, pengikatan fibril α-sinuklein ke membran mitokondria menyebabkan respirasi seluler yang rusak pada neuron primer, yang menyoroti hubungan dengan disfungsi mitokondria [ 73 ]. Akumulasi fibril Aβ menyebabkan stres mitokondria, yang dimanifestasikan oleh pembengkakan abnormal dan pembelahan mitokondria yang berlebihan. Perubahan morfologi ini disertai dengan gangguan fungsional yang semakin memperburuk ketidakseimbangan energi pada neuron [ 72 ]. Analisis kadar adenosin trifosfat (ATP) di bawah penghambatan selektif tahap-tahap spesifik metabolisme energi mengungkapkan pergeseran metabolik yang signifikan. Dalam kondisi disfungsi mitokondria, ada peningkatan β-oksidasi asam lemak dalam peroksisom bersama dengan intensifikasi glikolisis [ 72 ]. Dengan demikian, akumulasi fibril amiloid dapat memengaruhi sel-sel otak, khususnya astrosit, yang mengubah seluruh metabolisme energinya. Gangguan yang meluas ini pasti akan menyebabkan gangguan homeostasis otak, yang berpotensi mempercepat perkembangan penyakit neurodegeneratif.
Struktur seluler penting lainnya, di samping mitokondria, yang secara negatif dipengaruhi oleh fibril amiloid adalah retikulum endoplasma (ER) [ 74 , 75 ] (Gambar 2 ). ER memainkan peran penting dalam sintesis protein, pelipatan, dan transportasi. Fibril amiloid, melalui interaksinya dengan ER, mengganggu proses ini, yang mengarah pada akumulasi protein yang salah lipat di lumen ER. Kondisi ini, yang dikenal sebagai stres ER, mengaktifkan kaskade respons kompleks yang disebut respons protein tidak terlipat (UPR). Meskipun UPR awalnya ditujukan untuk memulihkan homeostasis, aktivasi jalur ini yang berkepanjangan dapat menyebabkan apoptosis [ 74 – 76 ]. Telah ditemukan juga bahwa akumulasi agregat intraseluler, tidak hanya di ER tetapi juga di sitoplasma, mengganggu asam ribonukleat (RNA), dan transportasi protein [ 77 ].
Akhirnya, penting untuk memperhatikan dampak fibril amiloid pada proses regulasi diri dan pembersihan sel, yang dalam kondisi normal membantu sel untuk mengatasi faktor stres [ 78 ] (Gambar 2 ). Fibril amiloid dilaporkan berkontribusi pada defek lisosomal [ 79 ], merekrut proteasom [ 80 ], sintase GMP-AMP siklik (cGAS) dan mengganggu autofagi—merupakan mekanisme kunci dari “pembersihan” sel yang bertanggung jawab atas degradasi organel dan agregat protein yang rusak [ 81 ]. Gangguan pada autofagi menghasilkan akumulasi komponen sel yang toksik, yang selanjutnya memperburuk stres sel dan berpotensi berkontribusi pada perkembangan penyakit neurodegeneratif.
Penting untuk menekankan bahwa gangguan pada tingkat organel individu tidak terjadi secara terpisah tetapi justru saling memperkuat satu sama lain, menciptakan respons stres seluler yang berjenjang. Salah satu komponen kunci dari respons ini adalah stres oksidatif. Induksi stres oksidatif merupakan mekanisme penting lain dari sitotoksisitas fibril amiloid. Agregat amiloid meningkatkan pembentukan ROS yang berlebihan [ 74 , 82 – 85 ], yang dapat terjadi melalui beberapa jalur, termasuk disfungsi mitokondria dan aktivasi nikotinamida adenin dinukleotida fosfat (NADPH) oksidase. Akumulasi ROS yang berlebihan menyebabkan kerusakan oksidatif pada asam deoksiribonukleat (DNA), lipid, dan protein, yang mengganggu fungsi normal struktur dan proses seluler [ 76 ]. Oksidasi lipid yang tidak terkontrol dapat mengganggu permeabilitas membran, memengaruhi fluiditas membran dan mengganggu fungsi protein transmembran [ 86 – 88 ]. Di dalam membran, lipid tak jenuh sangat sensitif terhadap serangan ROS pada ikatan rangkap asam lemak [ 89 ]. Kolesterol juga rentan terhadap oksidasi [ 90 ]. Modifikasi oksidatif peptida dan protein, terutama pada residu metionin, sistein dan tirosin, tidak hanya dapat mengganggu fungsinya, tetapi juga memodulasi proses agregasi protein (mengubah laju agregasi dan peralihan dari nukleasi primer ke sekunder) dan memengaruhi morfologi dan sitotoksisitas agregat yang terbentuk [ 91 – 98 ]. Produk aldehida dari peroksidasi lipid, 4-hidroksinonenal dan akrolein [ 99 ], dan produk oksidasi kolesterol yang mengandung aldehida reaktif terhadap residu sistein, lisin, atau histidin protein [ 100 , 101 ]. Modifikasi protein ireversibel oleh spesies ini dapat merangsang amiloidogenesis, seperti yang ditunjukkan dengan peptida β amiloid [ 101 ]. Pembentukan amiloid dan akumulasi ion logam di dalamnya berkontribusi terhadap pembentukan ROS, sehingga mendukung stres oksidatif [ 102 ]. Selain itu, struktur fibrilar, seperti yang telah ditunjukkan pada fibril β amiloid, dapat menghambat eliminasi lipid teroksidasi, yang akan meningkatkan peradangan dan persistensi peroksidasi lipid [ 103 ]. Pada gilirannya, produk reaktif dari oksidasi lipid dapat berkontribusi terhadap disfungsi mitokondria, ER, membran, dan kerusakan DNA lebih lanjut melalui ikatan silang [ 104 , 105]. Ketika produksi ROS membebani pertahanan antioksidan sel, atau ketika mekanisme pertahanan ini melemah, stres oksidatif kronis berkembang. Kondisi ini dapat memicu peradangan kronis, yang pada gilirannya memperburuk kerusakan sel, menciptakan lingkaran setan proses patologis [ 106 , 107 ].
2.2 Reaksi Berantai: Dari Kerusakan Organel hingga Aktivasi Sistem Imun
Yang penting, mekanisme kerusakan sel yang dijelaskan di atas tidak beroperasi secara terpisah dari reaksi imun yang diprakarsai oleh fibril amiloid; sebaliknya, mereka merupakan bagian integral dari proses kaskade yang kompleks. Bukti terbaru mengungkapkan hubungan mekanistik langsung antara kerusakan sel dan organel yang diinduksi amiloid dan aktivasi jalur siklik GMP-AMP sintase (cGAS)-STING, yang memicu respons imun bawaan [ 108 – 112 ]. Jalur cGAS–STING berfungsi sebagai komponen kunci dari pertahanan imun bawaan, yang memulai respons imun spektrum luas sebagai reaksi terhadap ancaman mikroba [ 113 ]. Namun, perlu dicatat bahwa cGAS juga dapat berinteraksi secara tidak normal dengan materi genetik yang berasal dari inang, seperti DNA ektopik dari nukleus atau mitokondria (mtDNA), ketika integritasnya terganggu [ 114 , 115 ]. Bahasa Indonesia: Ketika amiloid mengganggu integritas mitokondria atau nukleus, mereka memicu pelepasan pola molekuler terkait kerusakan (DAMP), termasuk DNA mitokondria (mtDNA) dan fragmen DNA nukleus. Asam nukleat ini, yang biasanya tersimpan dalam organel, menjadi terpapar secara abnormal pada sensor sitosol, seperti cGAS. Setelah mengikat DNA sitosol, cGAS mensintesis pembawa pesan kedua siklik GMP-AMP (cGAMP), yang mengaktifkan protein ER-resident stimulator of interferon genes (STING). Jalur cGAS-STING ini memulai kaskade pensinyalan hilir, termasuk aktivasi kinase pengikat TANK 1 (TBK1) dan faktor pengatur interferon 3 (IRF3), yang mengarah pada produksi interferon tipe I dan sitokin pro-inflamasi [ 109 , 112 , 116 , 117 ].
Hal ini kemungkinan mendasari peningkatan penelitian terkini yang menghubungkan regulasi abnormal jalur ini dengan berbagai kondisi neurologis (yang berhubungan dengan faktor-faktor termasuk akumulasi agregat amiloid) [ 112 , 118 ], meliputi penyakit Alzheimer [ 119 ], multiple sclerosis [ 120 ], penyakit Parkinson [ 121 ], ALS [ 122 ], penyakit Huntington [ 123 ], dan tauopathies [ 110 , 124 ].
Dengan mempertimbangkan konteks Bagian 1.1 , interaksi semacam itu dapat membentuk lingkaran umpan balik positif, di mana kerusakan sel primer yang diinduksi amiloid memulai jalur inflamasi, dan inflamasi yang dihasilkan meningkatkan produksi ROS dan memperburuk disfungsi organel, yang selanjutnya merangsang pelepasan DAMP DAMP [ 125 ]. Siklus kerusakan sel dan inflamasi yang berulang ini merupakan hubungan mekanistik penting dalam patologi amiloidosis, yang mengintegrasikan sitotoksisitas fibril langsung dan kerusakan jaringan yang dimediasi imun, sehingga menjelaskan sifat kondisi ini yang terus-menerus dan progresif.
2.3 Amplifikasi Peradangan: Dari Aktivasi Sistem Imun hingga Peradangan Kronis
Fibril amiloid dapat memicu respons imun tidak hanya secara tidak langsung, melalui kerusakan organel, tetapi juga secara langsung. Konformasi spasial amiloid yang atipikal berbeda secara signifikan dari protein asli, membuatnya secara eksplisit “terlihat” oleh sistem imun dan memulai serangkaian respons imun [ 126 – 131 ]. Secara khusus, respons imun telah diidentifikasi terhadap amiloid yang terbentuk dari Aβ, amilin pankreas, polipeptida amiloid pulau (IAPP), SOD1, α-sinuklein, huntingtin, dan lainnya [ 126 – 131 ]. Sistem imun mengidentifikasi fibril amiloid sebagai struktur patogenik melalui aktivasi reseptor pengenalan pola (PRR) spesifik, termasuk reseptor seperti tol (TLR) dan reseptor seperti NOD (NLR). Aktivasi reseptor ini memicu jalur pensinyalan intraseluler yang merangsang produksi sitokin dan kemokin pro-inflamasi, yang merekrut sel imun ke lokasi pengendapan amiloid [ 130 – 133 ]. Sistem komplemen memainkan peran penting dalam pengenalan fibril amiloid dan dapat diaktifkan setelah mengikat struktur fibrilar. Aktivasi komplemen memperkuat respons inflamasi dan merekrut fagosit [ 131 , 134 , 135 ]. Selain itu, sistem imun dapat menghasilkan antibodi spesifik yang menargetkan isoform amiloid, mengopsonisasi mereka untuk pembersihan selanjutnya oleh sel imun [ 136 – 138 ]. Namun, respons imun terhadap endapan amiloid seringkali tidak efektif dan dapat mengakibatkan konsekuensi yang merugikan [ 134 ]. Ketika sistem imun gagal membersihkan endapan amiloid secara menyeluruh—yang degradasinya secara menyeluruh, seperti yang akan ditunjukkan di bagian berikut, masih menjadi masalah yang belum terselesaikan—peradangan kronis dapat terjadi [ 135 , 138 , 139 ]. Kondisi ini ditandai dengan aktivasi sel imun yang berkelanjutan dan produksi mediator pro-inflamasi yang terus menerus, yang selanjutnya memperburuk kerusakan pada organ dan jaringan [ 140 – 142 ].
Untuk menyimpulkan bagian ini, kami ingin mengklarifikasi, mengapa meskipun efek amiloid tersebar luas pada kompartemen seluler yang berbeda, penyakit neurodegeneratif yang berbeda dengan gejala yang unik muncul. Intinya adalah bahwa meskipun agregat amiloid dapat mengganggu beberapa organel—seperti mitokondria, ER, lisosom, dan kompleks Golgi—gangguan ini tidak terjadi secara seragam di semua jenis sel atau area otak. Artinya, kerentanan khusus populasi neuronal tertentu, perbedaan dalam pemrosesan amiloid, dan keterlibatan respons imun yang berbeda dan peradangan yang terkompartementalisasi berkontribusi pada spesifisitas penyakit. Selain itu, sementara patologi amiloid merupakan fitur umum, efek hilir—seperti patologi tau, disfungsi sinaptik, dan neuroinflamasi—dapat berbeda dalam waktu, intensitas, dan lokalisasinya, yang mengarah pada presentasi klinis yang berbeda. Dengan demikian, keberadaan beberapa penyakit mencerminkan interaksi yang rumit antara toksisitas amiloid dengan konteks molekuler dan seluler spesifik penyakit, termasuk lingkungan seluler spesifik, latar belakang genetik, dan respons imun, yang mengakibatkan profil gejala yang berbeda-beda dan bukan sekadar kaskade sederhana di mana satu penyakit memicu penyakit lain atau di mana semua patologi merupakan efek samping dari satu proses tunggal.
3 Tantangan dalam Melawan Amiloid
3.1 “Keabadian”: Ilusi Kehancuran dan Kelahiran Kembali dari Abu
Meskipun telah ada upaya selama bertahun-tahun untuk memerangi amiloidosis, masih belum ada metode yang efektif dan aman untuk penghancuran total endapan amiloid. Seperti yang disebutkan sebelumnya, fibril amiloid telah lama dianggap sebagai agregat yang sangat tahan terhadap degradasi. Stabilitasnya dikaitkan dengan konformasi silang-β yang unik, di mana lembaran-β tersusun tegak lurus terhadap sumbu panjang fibril, membentuk struktur yang rapat. Ketahanan tinggi terhadap faktor eksternal ini dipastikan oleh banyaknya ikatan hidrogen yang terbentuk antara tulang punggung peptida dari untai-β yang berdekatan, serta interaksi antara rantai samping residu asam amino dalam polipeptida tetangga, seperti jembatan garam, interaksi hidrofobik, dan interaksi penumpukan [ 143 ].
Namun, penelitian baru-baru ini menunjukkan bahwa fibril amiloid memang rentan terhadap degradasi, meskipun sering tanpa disosiasi menjadi subunit monomerik. Beberapa faktor telah diidentifikasi yang, melalui mekanisme yang berbeda dan dengan berbagai tingkat efisiensi, dapat membongkar agregat patogen ini [ 144 ]. Ini termasuk kompleks chaperone disaggregase [ 144 ] dan berbagai enzim: matrix metalloproteinase (MMPs) [ 145 – 149 ], disintegrin dan protein yang mengandung domain metalloproteinase adm-2 [ 150 ], tripsin [ 151 – 153 ], high temperature requirement a serine peptidase 1 (HTRA1) [ 154 ] dan protease lisosomal seperti cathepsin [ 155 – 162 ]. Menariknya, ROS memainkan peran paradoks dalam degradasi amiloid. Di satu sisi, fibril amiloid dapat meningkatkan pembentukan ROS, sehingga memperburuk stres oksidatif dalam jaringan, seperti yang dibahas dalam Bagian 2.1 dari tinjauan ini. Di sisi lain, ROS dapat mendegradasi agregat amiloid dengan mengoksidasi residu asam amino utama [ 163 , 164 ]. Proses ini dapat menyebabkan fragmentasi rantai polipeptida dan pembentukan ikatan silang baru. Akibatnya, destabilisasi struktur fibril [ 165 ], fragmentasi [ 96 ] dan bahkan disosiasi menjadi bentuk monomerik dapat terjadi [ 166 ]. Diperkirakan bahwa mekanisme ini melibatkan gangguan interaksi hidrofobik [ 166 , 167 ], yang penting untuk menjaga integritas inti fibrilar. Selain itu, faktor fisikokimia lainnya dapat berkontribusi terhadap degradasi amiloid matang, termasuk tekanan mekanis [ 168 – 172 ], fluktuasi suhu [ 173 – 179 ], perawatan ultrasonik [ 180 – 184 ], dan penerapan agen denaturasi [ 151 , 185 – 187 ], deterjen ionik [ 188 – 190 ], dan pelarut organik [ 191 – 194 ].
Meskipun terdapat berbagai mekanisme degradasi, depolymerisasi lengkap fibril amiloid menjadi subunit monomerik tetap menjadi tantangan yang berat [ 195 ]. Akan tetapi, bahkan ketika teknik pencitraan menunjukkan penghancuran fibril yang efektif, penghentian faktor degradasi sering kali mengakibatkan penyusunan kembali faktor-faktor tersebut (Gambar 3 ). Fenomena ini dapat dikaitkan dengan keberadaan setidaknya sejumlah kecil “benih” amiloid di antara produk degradasi—fragmen fibril pendek yang mempertahankan strukturnya dan kapasitas untuk memanjang, sehingga mendorong repolymerisasi. Bahkan sejumlah kecil benih ini, yang sering kali tidak terdeteksi oleh metode konvensional, memiliki potensi yang cukup untuk memulai kembali proses fibrilogenesis [ 178 , 185 , 196 ].
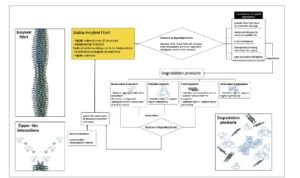
GAMBAR 3
Buka di penampil gambar
Presentasi PowerPoint
Regenerasi fibril amiloid setelah degradasi. Gambar tersebut menunjukkan kemampuan fibril amiloid yang luar biasa untuk memulihkan strukturnya bahkan setelah degradasi yang ekstensif. Berbagai faktor degradasi, termasuk protease, kompleks chaperone, ultrasonikasi, suhu, tekanan, ROS, dan deterjen ionik, mengganggu fibril amiloid, sering kali menghasilkan fragmen minimal. Degradasi dapat terjadi melalui destabilisasi berbagai gaya intramolekul, termasuk interaksi hidrofobik, hidrogen, ionik, dan kovalen (misalnya, melalui proteolisis atau oksidasi). Berbagai mode gangguan fibril menghasilkan pembentukan produk degradasi amiloid yang berbeda [ 144 ]: monomer terdenaturasi, oligomer amiloid, fragmen fibril, dan agregat yang terdestrukturisasi. Beberapa dari produk ini dapat memulihkan struktur asli fibril setelah menghilangkan efek denaturasi, karena fragmen fibril dengan struktur beta silang yang utuh bertindak sebagai benih untuk fibrilogenesis yang diperbarui. Repolymerisasi fibril didorong oleh interaksi seperti ritsleting dengan benih protein monomerik terlarut (yang tersisa setelah degradasi fibril atau yang baru disintesis) [ 197 ].
Kemampuan fibril amiloid yang luar biasa untuk mereplikasi diri setelah kerusakan yang nyata menyoroti kompleksitas dalam memerangi amiloidosis dan perlunya strategi komprehensif yang tidak hanya menargetkan degradasi fibril yang terakumulasi dalam tubuh tetapi juga mencegah pembentukannya dengan menghilangkan benih dan/atau menghambat pemanjangan dan nukleasi sekunder pada matriks ini.
3.2 “Perkalian”: Potong Satu Fibril, dan Banyak yang Akan Menggantikannya
Seperti yang dicatat dalam Bagian 3.1 , penghancuran amiloid yang hampir tuntas, sambil mempertahankan sejumlah kecil benih, dapat mengakibatkan “kelahiran kembali” mereka dari abu. Namun, konsekuensi yang lebih serius dari degradasi amiloid adalah fragmentasi mereka. Sekilas, pengurangan ukuran agregat amiloid besar seharusnya memfasilitasi pembersihan efektif mereka dari tubuh, sehingga mengurangi beban amiloid. Namun, studi terperinci tentang proses fragmentasi fibril telah mengungkap efek merugikannya.
Fragmentasi amiloid dapat terjadi secara spontan [ 198 ] dan/atau di bawah pengaruh berbagai faktor biologis (seperti protease mirip tripsin, chaperone, enzim lisosomal, ROS, dll., lihat Bagian 3.1 ) [ 144 , 199 ], serta diinduksi secara artifisial dalam kondisi laboratorium, misalnya, melalui perawatan ultrasonik [ 180 – 184 ]. Selama fragmentasi fibril amiloid, agregat besar dibagi menjadi fragmen yang lebih kecil yang serupa dalam struktur dan sifat dengan amiloid utuh (Gambar 4 ). Dengan demikian, fitur utama fragmentasi amiloid adalah bahwa hal itu sering terjadi tanpa mengganggu struktur β-sheet yang teratur yang merupakan karakteristik inti amiloid. Ini berarti bahwa fragmen yang dihasilkan mempertahankan kemampuan untuk berfungsi sebagai benih untuk agregasi protein lebih lanjut. Masing-masing fragmen ini berfungsi sebagai matriks untuk penempelan monomer protein bebas, sehingga mempercepat proses fibrilogenesis secara signifikan [ 50 , 183 , 200 – 202 ].
Detailnya ada di keterangan setelah gambar
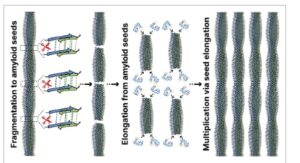
GAMBAR 4
Buka di penampil gambar
Presentasi PowerPoint
Amplifikasi fibril amiloid setelah fragmentasi. Selama fragmentasi fibril amiloid, agregat besar dibagi menjadi subunit yang lebih kecil tanpa mengganggu struktur lipatan β yang teratur yang menjadi ciri inti amiloid. Setiap fragmen ini menyediakan matriks untuk pelekatan monomer protein bebas (pelekatan dimungkinkan dari dua ujung), yang secara signifikan mempercepat proses fibrilogenesis, menyebabkan peningkatan proses inflamasi, stres oksidatif, dan disfungsi organel sel.
Mekanisme amplifikasi fibril amiloid melalui produk fragmentasi dapat dianggap sebagai suatu bentuk proses autokatalitik. Misalnya, perawatan ultrasonik pada amiloid matang meningkatkan konsentrasi “ujung bebas” fibril, yang menyebabkan peningkatan eksponensial dalam laju perlekatan monomer [ 50 , 183 , 200 – 202 ]. Dari perspektif patogenesis, peningkatan jumlah agregat amiloid yang dihasilkan dari fragmentasi dapat memperburuk proses inflamasi, stres oksidatif, dan disfungsi organel seluler. Secara kolektif, faktor-faktor ini berkontribusi pada percepatan perkembangan penyakit dan penurunan gambaran klinis.
Dengan demikian, kecenderungan tinggi fibril amiloid untuk mengalami fragmentasi menyoroti kompleksitas pengembangan strategi yang efektif untuk memerangi amiloidosis dan perlunya pendekatan komprehensif yang menangani semua aspek sifat dan toksisitas fibril amiloid. Dapat dihipotesiskan bahwa pemanfaatan fragmentasi untuk tujuan terapeutik dapat dilakukan jika dikombinasikan dengan metode pengobatan amiloid lain yang menghambat potensi penyebarannya.
3.3 “Kolonisasi” Organisme: Perkembangbiakan yang Tidak Terkendali
Kemampuan fibril amiloid untuk meningkat jumlahnya dengan cepat melalui fragmentasi dan replikasi diri tidak hanya menyebabkan pertumbuhan agregat amiloid yang seperti longsoran (lihat Bagian 2.2 ) tetapi juga penyebaran deposit amiloid yang cepat ke seluruh tubuh, sehingga secara efektif memindahkan patologi ke berbagai sel, jaringan dan organ [ 203 , 204 ] (Gambar 5 ).
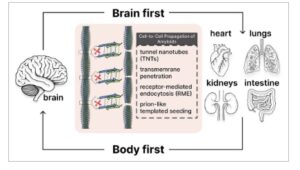
GAMBAR 5
Buka di penampil gambar
Presentasi PowerPoint
Perambatan fibril amiloid setelah fragmentasi. Setelah fragmentasi, baik spontan maupun yang disebabkan oleh pengaruh eksternal, benih fibrilar, seperti agen infeksius, menyebar dengan cepat dari jaringan dan organ yang terkena ke organ yang sehat. Mekanisme molekuler utama yang mendasari transmisi fibril amiloid antarsel meliputi: tunneling nanotubes (TNTs), penetrasi transmembran langsung, endositosis yang dimediasi reseptor (RME), dan pembenihan pola seperti prion. Penyebaran amiloid antara organ yang berbeda diilustrasikan oleh contoh penyakit Parkinson, di mana dua pola penyebaran α-sinuklein yang berbeda telah diidentifikasi: hipotesis “tubuh pertama” dan “otak pertama” (lihat teks untuk penjelasannya). Setelah menembus organ yang tidak terpengaruh, benih amiloid memulai agregasi protein asli, yang menyebabkan penskalaan pusat agregasi dan kerusakan pada sel dan jaringan yang sehat.
Seperti diketahui, mekanisme utama penyebaran fragmen amiloid adalah transfer antar sel [ 37 , 39–41 , 205 ]. Selain itu, praktik klinis telah melaporkan kasus penularan amiloid iatrogenik antara individu, yang terjadi melalui instrumen bedah yang terkontaminasi, transplantasi organ, atau suntikan ekstrak medis [ 206–208 ] . Begitu berada di jaringan dan organ yang sehat, fragmen amiloid bertindak serupa dengan agen infeksius . Mereka memulai agregasi protein asli, yang berfungsi sebagai benih di mana endapan amiloid baru terbentuk [ 41 , 50 , 144 , 151 , 152 , 183 , 185 , 200–202 , 205 , 209 , 210 ] .
Perambatan amiloid ke seluruh organisme menunjukkan kompleksitas yang luar biasa tidak hanya dalam jangkauannya tetapi juga dalam arahnya dan mekanisme molekuler yang mendasarinya. Bukti terbaru telah menyempurnakan pemahaman kita tentang proses ini, khususnya pada gangguan neurodegeneratif seperti penyakit Parkinson, di mana dua pola berbeda dari perambatan α-sinuklein telah diidentifikasi—hipotesis “tubuh-pertama” versus “otak-pertama” [ 211 , 212 ]. Pada subtipe “tubuh-pertama”, agregasi amiloid dimulai di jaringan perifer, khususnya sistem saraf enterik, dan kemudian naik ke sistem saraf pusat [ 212 – 215 ]. Bukti yang berkembang mendukung model ini: injeksi fraksi α-sinuklein ke dinding usus tikus mengakibatkan pengangkutan agregat ini bergantung waktu di sepanjang saraf vagus ke nukleus motorik dorsal vagus di batang otak [ 213 ]. Selain itu, α-synuclein agregat abnormal telah terdeteksi di saraf enterik sebelum kemunculannya di otak, menunjukkan bahwa racun yang berasal dari usus dan perubahan mikrobiota usus dapat memicu amiloidisasi α-synuclein [ 216 , 217 ]. Hal ini selanjutnya diperkuat oleh pengamatan klinis bahwa gejala non-motorik seperti sembelit dan gangguan perilaku tidur gerakan mata cepat sering mendahului manifestasi motorik selama bertahun-tahun atau bahkan puluhan tahun [ 218 – 220 ].
Sebaliknya, subtipe “otak-pertama” dimulai dengan agregasi protein di otak, yang berpotensi turun ke jaringan perifer di kemudian hari dalam perkembangan penyakit [ 211 , 221 ]. Studi primata non-manusia menunjukkan bahwa propagasi dua arah melalui sumbu usus-otak dapat terjadi, dengan sirkulasi sistemik berfungsi sebagai rute untuk transfer agregat α-sinuklein patogenik dari usus ke otak dan otak ke usus [ 216 ]. Pola propagasi dua arah yang serupa telah diamati pada penyakit Alzheimer, di mana patologi Aβ dapat berasal dari sistem saraf pusat atau jaringan perifer [ 222 – 224 ], dengan gangguan metabolik sistemik yang berpotensi mempercepat patologi amiloid sentral [ 225 ]. Pola propagasi yang berbeda ini mungkin mendasari heterogenitas dalam presentasi klinis, tingkat perkembangan, dan respons pengobatan yang diamati di antara pasien dengan diagnosis yang sama [ 211 , 226 ].
Pada tingkat molekuler, beberapa mekanisme khusus memfasilitasi transmisi fibril amiloid dari sel ke sel. Penetrasi transmembran langsung memungkinkan fibril amiloid melewati membran plasma dengan mendistorsi struktur lapisan lipid [ 7 , 59 ], difasilitasi oleh struktur β silang amiloid dan daerah hidrofobik yang terekspos. Endositosis yang dimediasi reseptor merupakan jalur utama lain, di mana fibril mengikat reseptor permukaan sel spesifik seperti proteoglikan heparan sulfat (HSPG) [ 227 ], gen aktivasi limfosit 3 (LAG3) [ 228 ], dan TLR [ 229 , 230 ], kemudian keluar dari kompartemen endosomal ke dalam sitosol [ 231 , 232 ]. TNT, jembatan membran tipis yang menghubungkan sel-sel yang jauh, menyediakan saluran langsung untuk transfer fibril amiloid antar sel, yang memungkinkan mereka untuk melewati lingkungan ekstrasel [ 64 – 66 , 233 ]. Struktur ini sangat lazim dalam kondisi stres seluler yang terkait dengan penyakit neurodegeneratif [ 234 – 237 ]. Proses penyemaian pola seperti prion, di mana fibril amiloid yang diinternalisasi menginduksi perubahan konformasi pada protein asli dengan kesetiaan spesifik strain yang luar biasa [ 203 , 238 – 240 ], menjelaskan berbagai manifestasi klinis dan patologis yang diamati pada amiloidosis.
Sifat-sifat amiloid yang dijelaskan tidak hanya menjelaskan mekanisme patogenik amiloidosis sistemik tetapi juga menggarisbawahi perlunya strategi diagnostik dan terapi komprehensif yang ditujukan untuk mencegah penumpukan amiloid di otak dan menekan penyebaran amiloid sistemik serta kemampuannya untuk memengaruhi jaringan baru.
Meskipun ada kompleksitas dalam memerangi amiloid yang dibahas di atas—yang terkait dengan “keabadian” mereka, “perkalian” yang tidak terkendali, dan kapasitas untuk “kolonisasi” di seluruh organisme—penting untuk mengakui kemajuan yang dicapai dalam beberapa tahun terakhir dalam mengembangkan agen anti-amiloid terapeutik. Secara khusus, kemajuan terbaru dalam terapi penyakit Alzheimer melibatkan antibodi monoklonal seperti Aducanumab, Lecanemab, dan Donanemab [ 241 , 242 ], yang menargetkan agregat Aβ dan telah menunjukkan kemampuan untuk memperlambat penurunan kognitif pada tahap awal penyakit dalam uji klinis. Namun, meskipun ada langkah maju yang signifikan ini, tantangan penyakit Alzheimer sayangnya masih belum terselesaikan. Tantangan yang signifikan tetap berupa permeabilitas antibodi yang rendah melintasi sawar darah-otak (BBB), yang membatasi potensi terapeutiknya dan menyoroti perlunya inovasi lebih lanjut dalam penghantaran obat [ 243 ]. Lebih jauh lagi, penerapan antibodi ini telah dikaitkan dengan efek samping potensial, yang sering disebut sebagai kelainan pencitraan terkait amiloid (ARIA), seperti edema otak, mikrohemoragi, efusi, hemoragi, dan atrofi otak [ 18 , 244 – 246 ]. Masuk akal bahwa beberapa efek samping ini, bersamaan dengan kegagalan translasi obat anti-amiloid lainnya yang belum memenuhi titik akhir primer dalam uji klinis tahap akhir, mungkin terkait, setidaknya sebagian, dengan konsekuensi yang timbul dari upaya degradasi amiloid, seperti yang dijelaskan dalam bagian ini (misalnya, degradasi tidak lengkap yang mengarah ke fragmen, oligomer, atau struktur yang berpotensi berbahaya).
4 “Ancaman Tersembunyi” dari Amiloid
4.1 “Transformasi”: Restrukturisasi dan Perubahan Properti
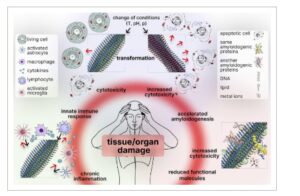
Kecenderungan fibril amiloid untuk terfragmentasi bukanlah satu-satunya karakteristik yang meningkatkan patogenisitasnya. Fibril amiloid menunjukkan polimorfisme yang luar biasa—kemampuan untuk menunjukkan berbagai macam struktur, sifat, dan, akibatnya, sitotoksisitas sebagai respons terhadap faktor endogen dan eksogen. Kemampuan untuk “bertransformasi” ini tidak hanya mempersulit pemahaman patogenesis amiloidosis tetapi juga membuat amiloid menjadi pemain yang lebih berbahaya dalam neurodegenerasi dan proses patologis lainnya di dalam tubuh.
Sejumlah faktor eksogen memainkan peran penting dalam transformasi struktural fibril amiloid [ 144 , 247-251 ] . Misalnya, perubahan pH lingkungan, peningkatan suhu, tekanan, dan rangsangan eksternal lainnya dapat memulai transformasi amiloid [ 151 , 152 , 173 , 188 , 247-253 ] (Gambar 6 , bagian atas). Perubahan-perubahan ini, pada gilirannya, dapat mengubah sifat-sifatnya secara signifikan dengan mengganggu interaksi lemah yang menstabilkan struktur fibril, sehingga memodifikasi kerentanannya terhadap agen pendegradasi [ 173 , 252 , 253 ], dan memodulasi interaksinya dengan molekul dan sel lain [ 254-261 ] . Penting untuk dicatat bahwa pembentukan produk degradasi yang tidak lengkap dari endapan amiloid dengan struktur yang berubah, yang diakibatkan oleh berbagai pengaruh eksogen , juga dapat dianggap sebagai bentuk “ transformasi” amiloid. Meskipun produk degradasi ini tidak memiliki struktur yang identik dengan fibril matang yang utuh, mereka mungkin masih mempertahankan toksisitasnya dan berkontribusi pada perkembangan proses patologis [ 144 , 185 , 262 ]. Selain itu, banyak penelitian telah menunjukkan bahwa “transformasi” tersebut bahkan dapat meningkatkan sitotoksisitas amiloid dibandingkan dengan fibril utuh [ 151 , 152 , 188 ] (Gambar 6 , bagian atas). Secara khusus, telah ditunjukkan bahwa dalam kondisi netral interaksi amiloid dengan protein syok panas kecil alfa-B-kristalin menyebabkan “melonggarnya” dan “gangguan” fibril, yang pada gilirannya, meningkatkan sitotoksisitasnya [ 188 ]. Karena alfa-B-kristalin hanya menyebabkan terganggunya fibril amiloid tanpa mengurangi ukurannya atau menghasilkan subunit oligomerik, maka peningkatan dampak patogenik amiloid setelah paparan pada kelangsungan hidup sel dapat dijelaskan oleh transformasi struktural fibril (termasuk perubahan pada “lapisan bulu” fibril), yang pada gilirannya meningkatkan afinitas amiloid terhadap membran plasma.
GAMBAR 6
Buka di penampil gambar
Presentasi PowerPoint
“Ancaman tersembunyi” dari amiloid. Perubahan kondisi lingkungan (seperti pH, suhu, dan tekanan) dan faktor eksternal lainnya dapat memicu transformasi amiloid. Dalam kebanyakan kasus, hal ini secara signifikan memengaruhi sifat-sifatnya (stabilitas, ketahanan terhadap degradasi, dan kemampuan untuk berinteraksi dengan sel), yang pada gilirannya dapat menyebabkan peningkatan sitotoksisitas dan kerusakan pada organ dan jaringan (ditunjukkan di atas). Konfigurasi spasial amiloid patologis berbeda dari protein asli, yang memicu respons imun. Namun, sistem imun tidak dapat menghilangkan endapan amiloid, yang menyebabkan perkembangan peradangan kronis. Kondisi ini ditandai dengan aktivasi sel imun yang konstan dan produksi mediator pro-inflamasi, yang selanjutnya memperburuk kerusakan organ dan jaringan (ditunjukkan di sebelah kiri). Plak amiloid, dengan mengumpulkan berbagai komponen molekuler seperti protein, lipid, ion logam, DNA, dan lainnya, tidak hanya “menonaktifkan” komponen-komponen ini tetapi juga memperoleh sifat patogenik baru (pertumbuhan yang dipercepat dan peningkatan stabilitas dan sitotoksisitas amiloid). Tindakan sitotoksik molekul yang “ditangkap” oleh fibril amiloid memperburuk dampak patologis amiloid, menciptakan pola gangguan kompleks yang melampaui pengaruh fibril amiloid yang terisolasi. Panah hitam menunjukkan pengaruh eksternal pada fibril amiloid, sedangkan panah merah menunjukkan efek yang diberikan oleh amiloid pada sel dan molekul di sekitarnya.
Dengan demikian, kemampuan amiloid untuk bertransformasi menekankan pentingnya mempertimbangkan tidak hanya fibril amiloid itu sendiri tetapi juga interaksinya dengan lingkungan mikro saat mempelajari patogenesis amiloidosis. Mengungkap mekanisme molekuler dan konsekuensi restrukturisasi amiloid membuka perspektif baru untuk mengembangkan metode pengobatan yang efektif. Penelitian dan pendekatan terapeutik di masa mendatang harus memperhitungkan sifat dinamis amiloid untuk memerangi tidak hanya bentuk awal fibril amiloid tetapi juga varian transformasinya yang berpotensi lebih berbahaya.
4.2 “Daya Tarik” dan “Patogenisasi” Spesies Sekitar: Kehilangan Fungsi dan Konsolidasi Patologis dengan Fibril Amiloid
Fibril amiloid menimbulkan ancaman bagi organisme tidak hanya karena sitotoksisitasnya yang tinggi, resistensi terhadap degradasi, perbanyakan cepat, dan penyebaran yang luas. Selama amiloidogenesis, protein asli mengalami perubahan konformasi, membentuk agregat fibrilar β-sheet yang khas. Bahkan “benih” amiloid pendek bertindak sebagai perangkap, menangkap molekul protein yang aktif secara fungsional dan menyebabkannya mengadopsi konformasi patologis [ 263 – 268 ]. Proses ini menyebabkan penipisan progresif kumpulan protein fungsional dalam sel, mengganggu proses biologis penting (Gambar 6 , kanan).
Selain itu, agregat amiloid berkontribusi pada penggabungan ke dalam struktur fibrilar hanya molekul protein target yang membentuk benih amiloid tetapi juga protein vital lainnya [ 269 – 272 ] (Gambar 6 , kanan). Mekanisme penyemaian silang ini secara signifikan memperluas jangkauan potensi gangguan yang disebabkan oleh amiloidogenesis, yang memengaruhi fungsi beberapa protein secara bersamaan. Saat agregat amiloid tumbuh, ketersediaan protein asli dalam sel berkurang dengan cepat. Akibatnya, seiring dengan perluasan autokatalitik endapan amiloid patologis, proses seluler penting yang bergantung pada fungsi normal protein ini terganggu.
Perlu juga dicatat bahwa rentang molekul yang terlibat dalam agregat amiloid tidak terbatas pada protein amiloidogenik yang secara langsung membentuk fibril [ 273 ]. Gugus amiloid mengandung berbagai komponen, termasuk ribuan protein inflamasi dan fungsional yang berbeda dalam bentuk non-fibrilar [ 274-279 ] , imunoglobulin [ 273 , 280 , 281 ], serta glikosaminoglikan [ 282 , 283 ] , lipid [ 284-286 ], ion logam [ 287-291 ], dan asam nukleat , terutama RNA [ 292-294 ] (Gambar 6 , kanan). Beberapa protein yang terkait dengan endapan memiliki epitop spesifik pada permukaan amiloid (misalnya, imunoglobulin ), tetapi sebagian besar pengikatan protein pada endapan bergantung pada interaksi nonspesifik [ 273-295 ] . Glikosaminoglikan (khususnya, HSPG yang ditemukan dalam deposisi Aβ) terlibat dalam interaksi elektrostatik langsung dan spesifik dengan peptida/protein pembentuk amiloid, yang berkontribusi pada perkembangan patologi amiloid [ 295 – 298 ]. Studi terbaru menunjukkan bahwa interaksi antara protein dalam agregat amiloid dan asam nukleat bersifat polivalen dan sebagian besar bersifat elektrostatik [ 293 , 294 ]. Meskipun demikian, ada bukti interaksi spesifik antara sekuens tertentu pada permukaan amiloid dan RNA [ 292 ]. Ion logam secara langsung berkoordinasi dengan residu histidin dari protein dan peptida amiloidogenik, yang memengaruhi agregasi dan nukleasi plak amiloid [ 287 ]. Akumulasi ion logam dalam plak amiloid dapat dimediasi oleh pengikatannya dengan protein terkait plak lainnya [ 298 ]. Ion logam juga dapat diikat secara spesifik oleh glikosaminoglikan dalam cara tetragonal [ 298 , 299 ]. Interaksi struktur lipid, yang sering berfungsi sebagai platform untuk perakitan amiloid, dengan protein dan peptida amiloidogenik melibatkan interaksi hidrofobik dan ikatan hidrogen [ 298 , 300 ].
Akumulasi berbagai komponen dalam amiloid menyebabkan hilangnya aktivitas fungsionalnya, yang dapat berkontribusi pada perkembangan berbagai patologi, termasuk yang tampaknya tidak terkait dengan amiloidosis. Secara khusus, interaksi RNA dengan amiloid [ 292 – 294 ] dapat mengganggu proses seluler normal di mana RNA memainkan peran kunci dalam membawa informasi genetik. Ini dapat memiliki konsekuensi serius untuk ekspresi gen, sintesis protein, dan jalur biokimia penting lainnya yang bergantung pada fungsi RNA yang tepat. Pengurangan kadar glikosaminoglikan fungsional mengganggu pembentukan matriks ekstraseluler. Dimasukkannya enzim seperti gliseraldehid-3-fosfat dehidrogenase (GAPDH) [ 295 , 300 , 301 ] dan enolase [ 278 , 279 , 302 ] ke dalam plak amiloid pada penyakit Alzheimer mengganggu glikolisis. Metabolisme lipid juga terganggu oleh ko-agregasi lipid dan endapan amiloid [ 284 – 286 ] yang berpotensi berkontribusi terhadap penurunan kognitif.
Pada saat yang sama penggabungan berbagai molekul ke dalam fibril amiloid, baik selama fibrilogenesis dan setelah pembentukan fibril selesai, juga dapat meningkatkan sifat patogenik dan meningkatkan sitotoksisitas amiloid, serta mempercepat amiloidogenesis (Gambar 6 , kanan). Misalnya, glikosaminoglikan dapat meningkatkan stabilisasi dan pertumbuhan fibril amiloid [ 282 , 283 ]. Proteoglikan juga dapat mempercepat pembentukan amiloid dan melindunginya dari degradasi proteolitik [ 255 ]. Hemoglobin, yang terdeteksi dalam plak amiloid pada penyakit Alzheimer, meningkatkan agregasi peptida amiloidogenik [ 303 ]. Ion logam [ 290 , 291 , 304 , 305 ] tidak hanya mempercepat agregasi struktur protein tetapi juga mengkatalisis pembentukan ROS setelah mengikat amiloid, sehingga meningkatkan stres oksidatif dalam sel [ 288 – 290 , 306 ]. Interaksi protein dan peptida amiloidogenik dengan komponen utama kaskade koagulasi dapat meningkatkan pembentukan trombin dan menyebabkan akumulasi fibrin patologis [ 307 ]. Hal ini, pada gilirannya, dapat berkontribusi terhadap disfungsi dan degenerasi neuronal pada penyakit Alzheimer dengan menginduksi neuroinflamasi atau mengganggu aliran darah otak [ 308 ]. Molekul RNA yang berasosiasi dengan amiloid [ 292 – 294 ] dapat berfungsi sebagai kofaktor dalam patogenesis amiloidosis, sehingga meningkatkan toksisitas agregat amiloid. Khususnya, agregat amiloid yang dimodifikasi oleh molekul yang direkrut dapat meningkatkan respons inflamasi. Misalnya, aktivasi mikroglia dan astrosit yang berkelanjutan telah terbukti mengakibatkan neuroinflamasi kronis [ 309 , 310 ].
Dengan demikian, plak amiloid, melalui akumulasi berbagai komponen molekuler, memperoleh sifat patogenik tambahan. Efek sitotoksik dari molekul yang diasingkan ini memperkuat pengaruh patologis amiloid itu sendiri, yang mengarah ke serangkaian disfungsi yang kompleks, yang tidak khas untuk struktur amiloid yang utuh. Ini berarti bahwa pengembangan strategi anti-amiloid harus ditujukan tidak hanya untuk menghambat interaksi protein amiloidogenik satu sama lain untuk mencegah fibrilogenesis tetapi juga untuk memblokir interaksi amiloid dengan molekul vital lainnya dalam tubuh, sehingga mempertahankan aktivitas fungsionalnya.
5. Kesimpulan
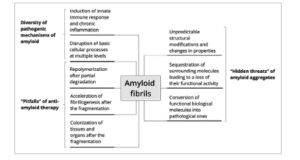
Dalam tinjauan ini, kami telah mensistematisasi dan menganalisis secara kritis data kontemporer tentang keterlibatan fibril amiloid dalam patogenesis gangguan neurodegeneratif dan berbagai penyakit lainnya (Gambar 7 ). Kami telah menunjukkan bagaimana amiloid merusak proses seluler dengan memengaruhi berbagai organel. Agregat protein ini memiliki kemampuan luar biasa untuk melakukan repolimerisasi setelah penghancuran dan dapat tumbuh dalam ukuran dan jumlah melalui fragmentasi. Amiloid menyebar ke seluruh tubuh, memperluas pengaruh patologisnya, dan di bawah kondisi eksternal, mereka dapat mengalami perubahan struktural yang meningkatkan toksisitasnya. Lebih jauh lagi, mereka dapat memicu peradangan kronis, memprovokasi respons imun yang berkepanjangan dan merusak. Amiloid menarik berbagai molekul, menghilangkan aktivitas fungsionalnya, dan kemudian mengisolasi molekul-molekul ini, mengubahnya menjadi agen patologis dengan sifat destruktif yang baru. Analisis kami menunjukkan bahwa amiloid menunjukkan potensi patogenik multifaset yang beroperasi pada berbagai tingkat organisasi biologis. Meskipun munculnya target terapi baru menyusul pergeseran fokus penelitian terkini terhadap oligomer dan protofibril, serta penyebab potensial penyakit neurodegeneratif lainnya, amiloid tetap menjadi subjek studi penting dalam biomedis modern.
GAMBAR 7
Buka di penampil gambar
Presentasi PowerPoint
Peran yang beragam dan “bahaya tersembunyi” dari fibril amiloid. Skema ini menggambarkan sifat fibril amiloid yang kompleks dan beragam, yang menunjukkan efek patogenik pada manusia. Aspek-aspek utamanya meliputi kemampuan adaptasi strukturalnya, kapasitasnya untuk mengganggu sel dan sistem imun, dan keterlibatannya dalam proses peradangan kronis.
Data yang dikumpulkan dan dianalisis dalam tinjauan ini membantu memahami asal-usul paradoks amiloid, memberikan wawasan mengapa fibril amiloid tetap menjadi tantangan yang terus-menerus meskipun telah dilakukan penelitian selama beberapa dekade. Karakteristik unik amiloid yang dijelaskan di sini menjelaskan kemanjuran terbatas dan efek samping dari obat-obatan pendegradasi amiloid komponen tunggal dan imunoterapi yang diusulkan untuk penyakit neurodegeneratif, di mana paparan berbagai enzim yang tidak terkontrol terhadap amiloid menyebabkan konsekuensi yang tidak dapat diprediksi. Temuan sistematis kami dapat membantu dalam pengembangan strategi terapi yang efektif dan aman untuk memerangi agregat protein ini sambil mempertimbangkan konsekuensi pengobatan yang potensial. Tinjauan tersebut, khususnya, menunjukkan bahwa tantangan signifikan ada di depan bagi para peneliti dan dokter, terutama dalam mengembangkan metode yang tidak hanya dapat menghancurkan fibril amiloid tetapi juga mencegah pembentukannya kembali. Kemampuan khas amiloid untuk meregenerasi diri, muncul kembali, dan beradaptasi dengan kondisi yang berbeda membuat pengembangan strategi terapi yang efektif menjadi sangat rumit.
Kami percaya bahwa solusi yang menjanjikan untuk amiloidosis mungkin terletak pada pendekatan gabungan yang melibatkan penargetan agregat patologis melalui integrasi beberapa mekanisme tindakan, yang berpotensi mengatasi keterbatasan dan efek samping dari pendekatan individual [ 311 ]. Secara khusus, strategi terapi masa depan harus fokus pada degradasi amiloid yang terkontrol secara menyeluruh daripada degradasi parsial, seperti yang diamati pada obat-obatan yang dikembangkan saat ini. Dengan tujuan ini, strategi terapi harus mengatasi beberapa tantangan utama:
Pengiriman yang ditargetkan: mencapai penetrasi BBB yang cukup dan spesifisitas jenis sel tetap menjadi rintangan utama untuk terapi anti-amiloid. Desain senyawa rasional dan memanfaatkan mekanisme transportasi endogen sangat penting untuk memastikan degradator mencapai target yang dituju dalam sistem saraf pusat. Kemajuan terkini mencakup pengembangan sistem pengiriman obat bertarget ganda dan penggunaan nanopartikel atau modifikasi ligan untuk meningkatkan persilangan BBB dan menargetkan daerah saraf atau jenis sel tertentu yang terlibat dalam patologi Alzheimer [ 312 – 315 ]. Ultrasonografi terfokus dalam kombinasi dengan terapi antibodi monoklonal juga menunjukkan harapan dalam membuka BBB sementara, sehingga meningkatkan akses obat ke area otak yang terkena [ 316 ]. Namun, strategi ini harus menyeimbangkan kemanjuran dengan keamanan, karena efek di luar target dan risiko ARIA tetap menjadi perhatian yang signifikan [ 317 , 318 ].
Meminimalkan zat antara yang beracun: selama degradasi, terdapat risiko menghasilkan agregat mirip amiloid dengan struktur yang berubah atau spesies oligomerik yang larut, yang mungkin lebih neurotoksik daripada agregat yang utuh. Hal ini menggarisbawahi perlunya pendekatan yang mencegah akumulasi zat antara ini atau dengan cepat membersihkannya dari lingkungan seluler [ 311 , 317 ]. Antibodi terapeutik telah dikembangkan untuk mengikat dan menetralkan bentuk oligomerik ini secara selektif, sementara diagnosis dini berdasarkan biomarker dapat memungkinkan intervensi sebelum kerusakan ireversibel terjadi [ 318 ].
Mencegah repolymerisasi: strategi juga harus mempertimbangkan potensi fragmen yang terdegradasi untuk beragregasi ulang, yang memerlukan penghilangan yang efisien atau penghambatan jalur agregasi secara bersamaan. Ekspresi berlebihan enzim proteolitik dan antibodi spesifik telah dieksplorasi untuk mendorong penghilangan protein yang salah lipat, termasuk monomer yang berasal dari fibril. Di antara pendekatan yang paling menjanjikan adalah proteolysis-targeting chimeras (PROTACs), yang memanfaatkan sistem ubiquitin–proteasome untuk mendegradasi protein target secara selektif [ 18 ]. Banyak inhibitor—termasuk molekul kecil dan peptida—telah dikembangkan untuk mencegah pembentukan dan re-agregasi fibril, bertindak sebagai “perangkap” molekuler untuk monomer. Namun, banyak dari mereka masih menghadapi tantangan terkait farmakokinetik, kelarutan, dan stabilitas. Strategi seperti menggabungkan inhibitor ini dengan nanoliposom sedang diuji untuk meningkatkan sifat-sifatnya. Selain itu, domain BRICHOS rekombinan (Bri2, kondromodulin-1, dan protein pro-surfaktan C) telah muncul sebagai pendamping penghambat amiloid yang serbaguna, efektif melawan berbagai peptida dan protein amiloidogenik yang relevan dengan berbagai penyakit neurodegeneratif [ 243 ].
Perlu dicatat pula bahwa, mengingat mekanisme patogenik amiloid yang beraneka ragam yang dijelaskan dalam tinjauan ini, prioritas penelitian di masa mendatang harus diperluas melampaui degradasi/repolimerisasi amiloid untuk mencakup analisis terperinci tentang interaksinya dengan biomolekul vital lain dalam tubuh manusia. Investigasi semacam itu akan membangun dasar untuk mengembangkan pendekatan yang ditargetkan untuk menghambat interaksi ini, dengan demikian menetralkan efek merugikannya pada proses fisiologis. Studi-studi ini tidak hanya akan meningkatkan keamanan dan kemanjuran intervensi terapeutik dan meningkatkan hasil pasien tetapi juga memajukan pengembangan pendekatan untuk diagnosis dini penyakit neurodegeneratif dan penyakit terkait amiloid lainnya.